


Interpreting BLAST output

Here is a sample blast result (from BLAST on the NCBI site, using a tomato sequence as a query)
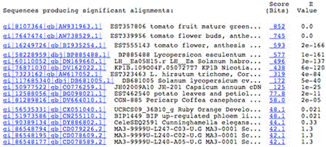
It is important to realize that there is no set cut-off which determines whether a match is considered significant or "similar enough" - this has to be set by the user.
In this example, the second "hit" is another EST sequence, but with exactly the same E-value as the self hit, implying that this is probably a redundant sequence - at the very least, its sequence matches the query over its full length.
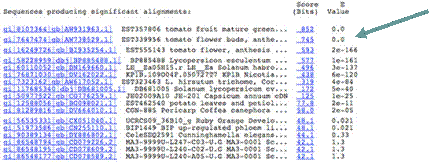
Further down the list is an EST from potato leaves (EST462540) with an E-value of -11. This would be considered as borderline similar. Any hits worse than this would generally not be considered significantly similar to the query sequence.
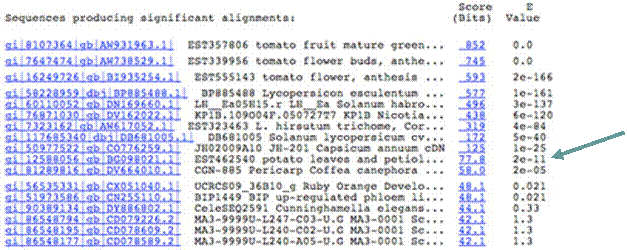
Clicking on the Score (bits) in the right column (highlighted in blue) brings up the detailed alignment. Below is the output from clicking on the score of the second tomato hit, which shows a perfect alignment at all the nucleotide positions.
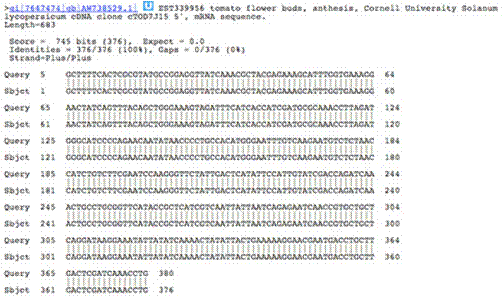
Below is the output of clicking on the much less significant potato hit. Only a very short stretch is present over which the sequences are aligned, and even then there are nucleotides which differ between the two sequences.


